CSIR-IHBT
IDIOPATHIC PULMONARY FIBROSIS INFORMATION PORTAL
(A Molecular Systems Database for IPF)
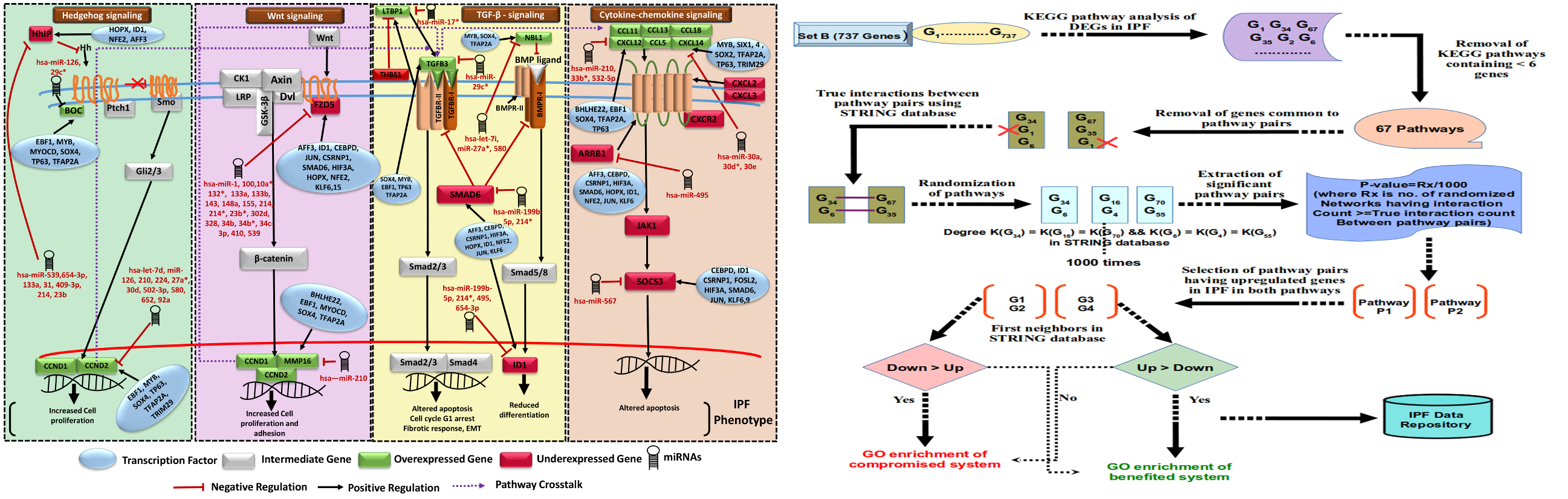
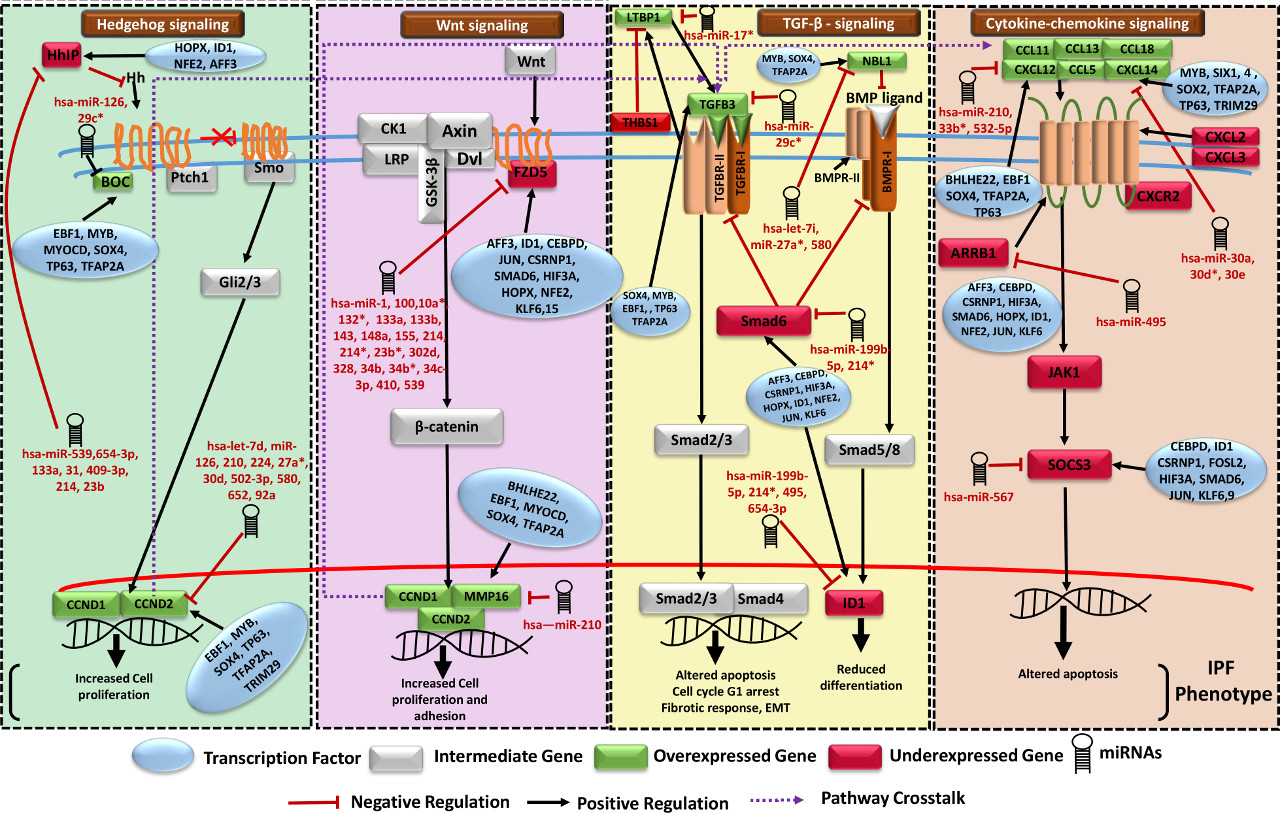
Hypothetical model of IPF regulatory network involving four crucial biological pathways
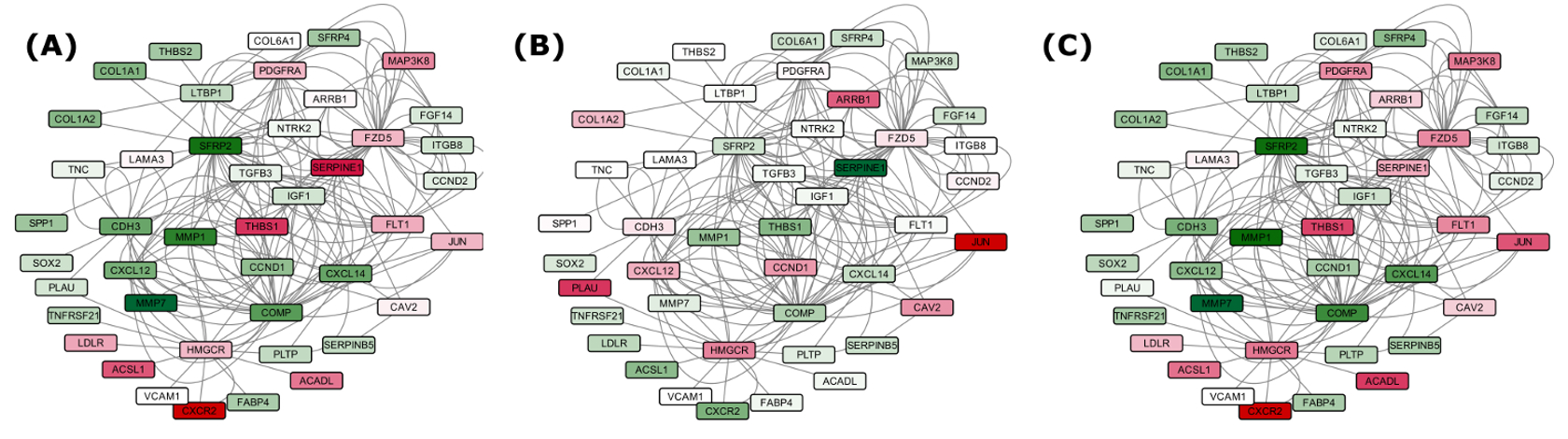
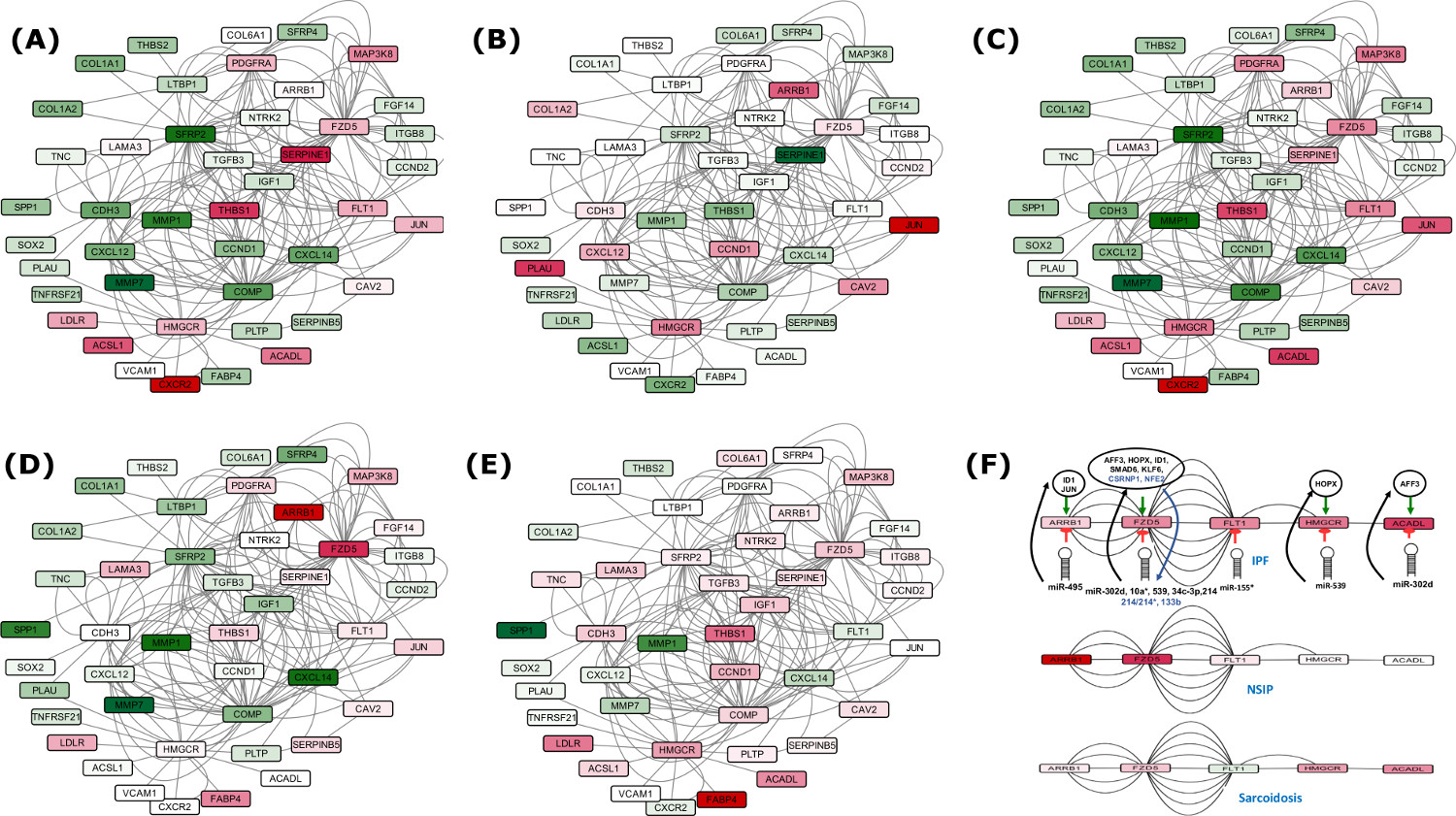
Disease specific comparison of DEG in Set A (39) with IPF, Sarcoidosis, NSIP.
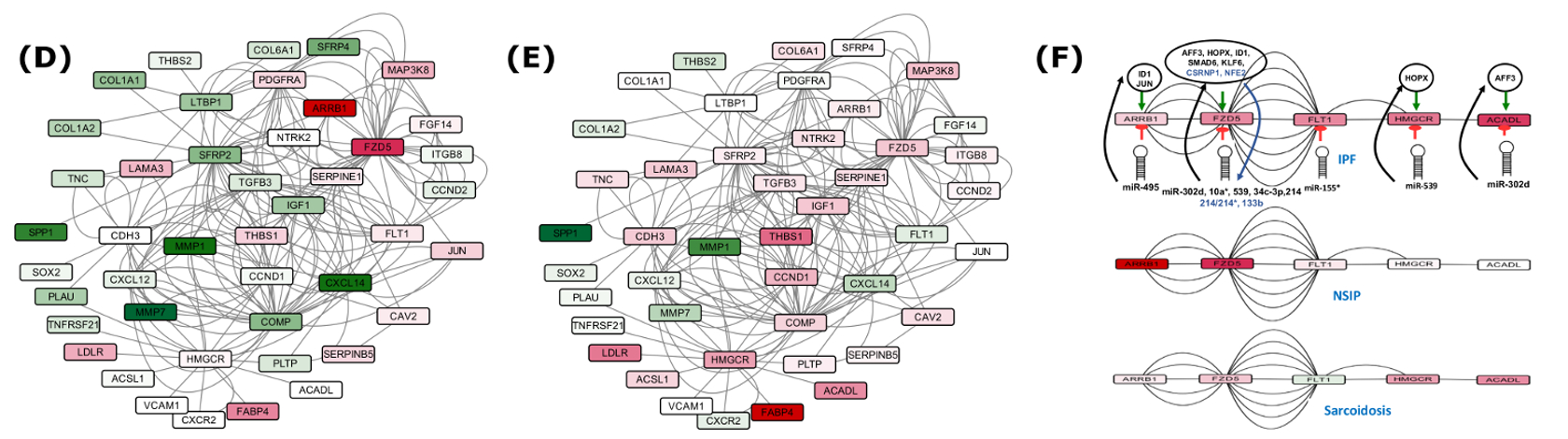
Five critical check points differentially downregulated exclusively in IPF contary to NSIP and Sarcoidosis.
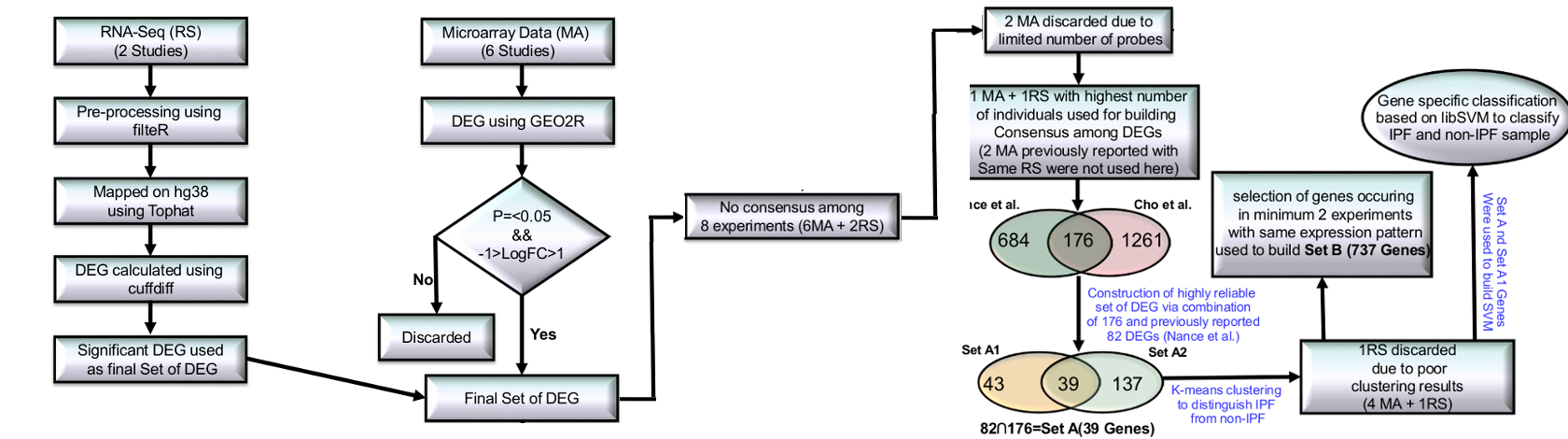
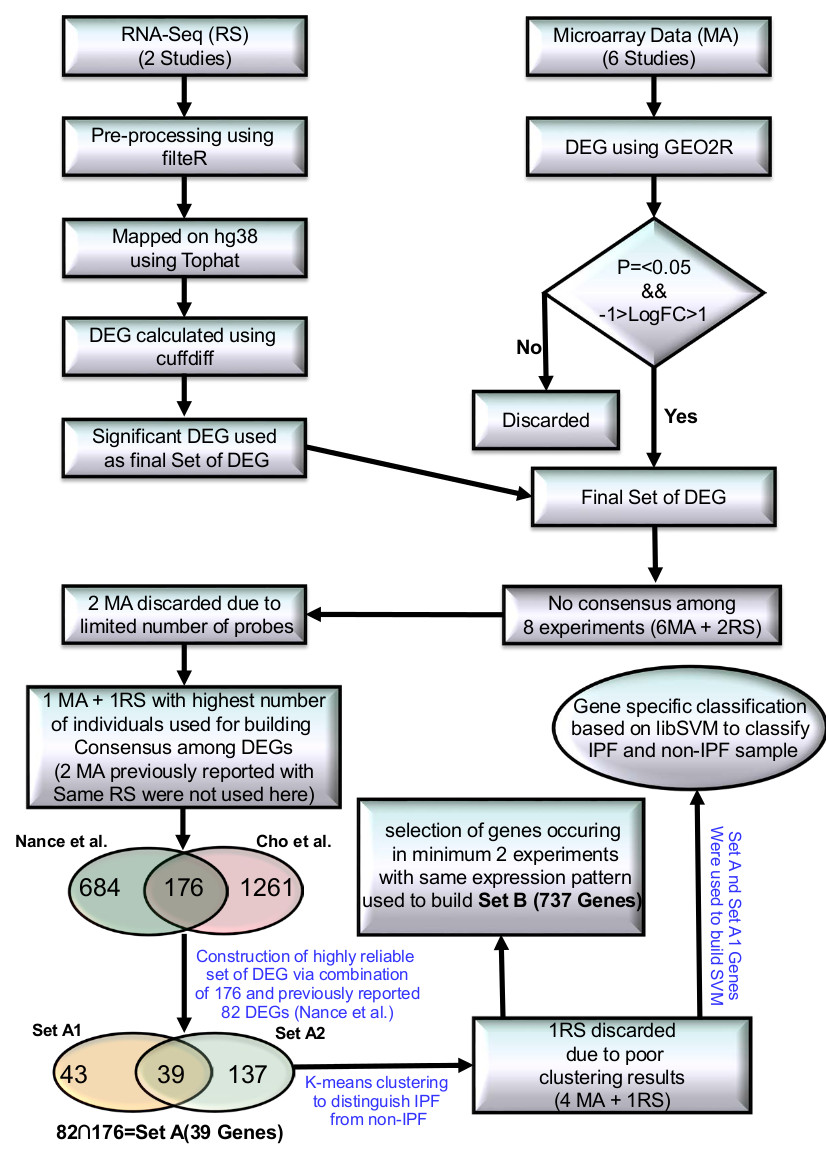
Protocol showing various steps of high-throughput data processing to build significant DEG sets.
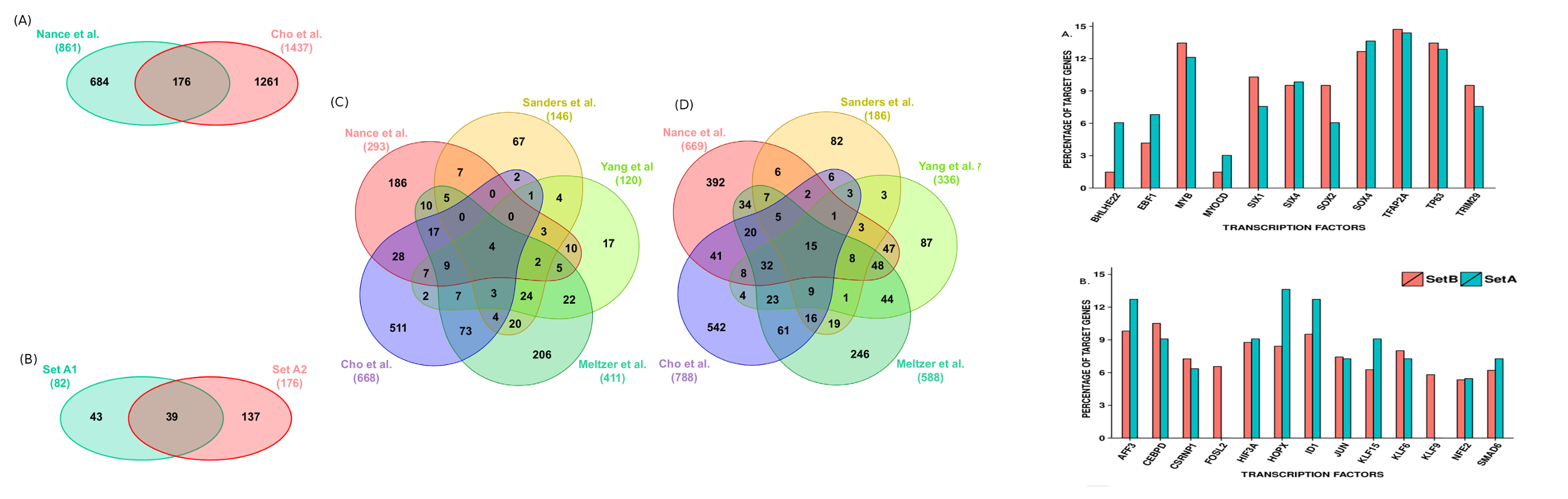
Overlap of expressed genes in different studies of IPF and ranking of transcription factor target genes.
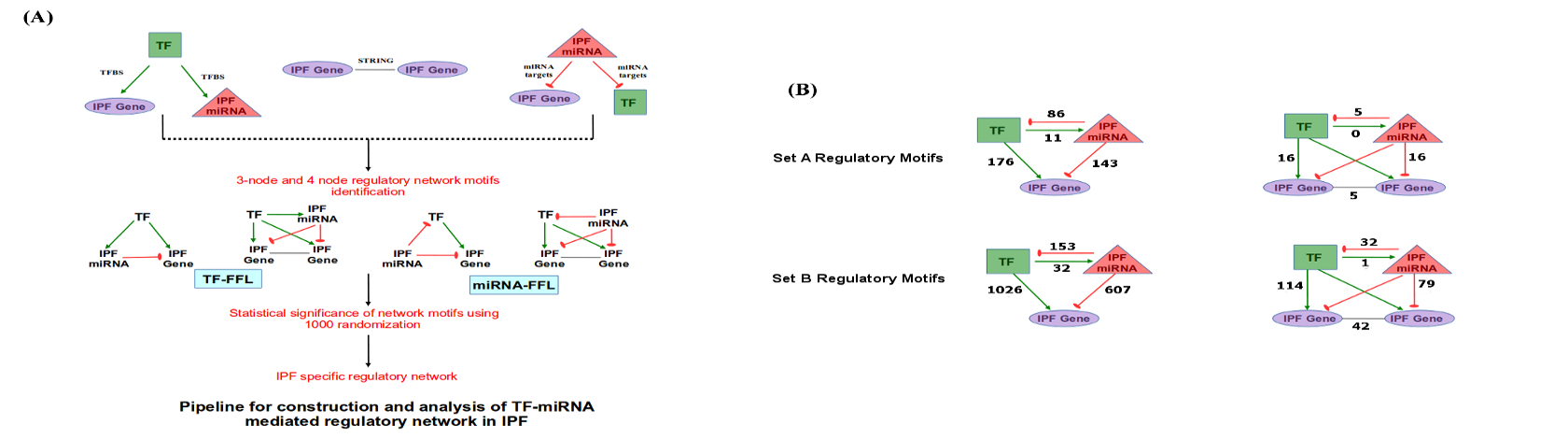
Pipeline for construction and analysis of TF-miRNA mediated regulatory network in IPF.
Quick search
Browse here
Chromosomal distribution of differential expressed genes in IPF
The Article:)
Idiopathic Pulmonary Fibrosis is an incurable progressive fibrotic disease of the lungs. We currently lack a systematic understanding of IPF biology and a systems approach may offer new therapeutic insights. Here, for the first time, a large volume of high throughput genomics data has been unified to derive the most common molecular signatures of IPF. A set of 39 differentially expressed genes (DEGs) was found critical to distinguish IPF. Using high confidence evidences and experimental data, system level networks for IPF were reconstructed, involving 737 DEGs found common across at least two independent studies.
This all provided one of the most comprehensive molecular system views for IPF underlining the regulatory and molecular consequences associated. 56 pathways crosstalks were identified which included critical pathways with specified directionality. The associated steps gained and lost due to crosstalk during IPF were also identified. A serially connected system of five crucial genes was found, potentially controlled by nine miRNAs and eight transcription factors exclusively in IPF when compared to NSIP and Sarcoidosis. Findings from this study have been implemented into a comprehensive molecular and systems database on IPF to facilitate devising diagnostic and therapeutic solutions for this deadly disease.
Idiopathic interstitial pneumonias (IIPs) are interstitial lung diseases with unidentified mechanism, distinguished by matrix deposition and alveolar epithelium disruption. Idiopathic pulmonary fibrosis (IPF) is a chronic fibrosing IIP that has no effective therapy with a mortality rate higher than many cancers; median survival from the time of diagnosis being about three years1. There is typically no response to general anti-inflammatory therapy such as glucocorticosteroids, which are effective in some IIPs such as non-specific interstitial pneumonia (NSIP). Antifibrotic agents such as Nintedanib and Pirfenidone were initially reported to be potentially beneficial, but the early promise has not been sustained2-3. Adenosine receptor antagonist based solutions have shown some limited promise4. While new targets have emerged, such as proinflammatory cytokine (IL-13)5, the negligible molecular systems information for IPF remains a problem. A gene expression level understanding of IPF is incomplete, with limited number of studies mostly microarray (MA) based6-11 and hardly a couple of RNA-seq studies12-13. Also, very few study has been done covering systems biological perspectives but not specific to IPF9. Limited MA based studies with non-coding RNAs have also been performed, but small RNA-seq studies are lacking14-16.
From a systems biology perspective, IPF offers both opportunities and challenges. The main challenge is a likely heterogeneity within IPF, which is a clinical diagnosis based on typical diffuse radiological or pathological findings that may be seen in a limited form in other IIPs or even aged lungs. Further, the IPF lung is itself heterogeneous. Thus there is likely to be substantial background noise, as evidenced by high variability between studies. This also presents an opportunity to systematically consolidate all these studies, using informatics to extract the underlining characteristic signatures for IPF. Such efforts are critical towards better molecular characterization of IPF that could lead to better diagnosis, classification and therapy.
From a systems biology perspective, IPF offers both opportunities and challenges. The main challenge is a likely heterogeneity within IPF, which is a clinical diagnosis based on typical diffuse radiological or pathological findings that may be seen in a limited form in other IIPs or even aged lungs. Further, the IPF lung is itself heterogeneous. Thus there is likely to be substantial background noise, as evidenced by high variability between studies. This also presents an opportunity to systematically consolidate all these studies, using informatics to extract the underlining characteristic signatures for IPF. Such efforts are critical towards better molecular characterization of IPF that could lead to better diagnosis, classification and therapy.
This is important because the therapeutic response is very different between different types of IIP and possibly within different subsets of IPF. There is also a need for unified information portal and database for molecular and systems biology of IPF beyond the few existing resources on expression data reporting that have limited scope (https://research.cchmc.org/pbge/lunggens/mainportal.html http://montgomerylab.stanford.edu/resources.html). The present study has been carried out considering these factors. The study attempts to unify the best available gene expression data for IPF, derive its core characteristics towards generating a computational model of IPF pathology and create a comprehensive state-of-the-art database dedicated to IPF research.
Data processing and analysis
Fourteen high throughput studies were included containing six IPF MAs, two RNA-seq , three miRNA MA, two MAs for Sarcoidosis41-42 and one MA of NSIP43 (Supplementary Table S12). All initial data from MA and RNA-seq based high throughput studies were collected from GEO and SRA. RNA-seq data was cleaned using filteR44. TOPHAT 2.1.0 was used for mapping RNA-seq reads across the human genome (hg38) with default parameters. The alignment results were saved in BAM format. CUFFLINK-CUFFDIFF tools were used for expression analysis for RNA-seq data. The work flow of IPF high-throughput data processing to generate high confidence gene sets is given in Figure 3. For Micro-array gene data, GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) tool containing R, Biobase46, GEOquery47 and limma48 packages was used with some basic changes in Rscript. Based on the information derived from the clustering of different gene sets with varying number of DEGs (detailed in the result section), set of feature genes was formed. The relative genome wide normalized expression values of these feature genes were used for vector transformational representation of each individual sample considered in this study. Features expression transformation into Z-score for each experimental dataset was done to bring the expression values on same scale irrespective of their platform and experimental design. The classification was implemented with 5-fold cross validation using LibSVM.
Fourteen high throughput studies were included containing six IPF MAs, two RNA-seq , three miRNA MA, two MAs for Sarcoidosis41-42 and one MA of NSIP43 (Supplementary Table S12). All initial data from MA and RNA-seq based high throughput studies were collected from GEO and SRA. RNA-seq data was cleaned using filteR44. TOPHAT 2.1.0 was used for mapping RNA-seq reads across the human genome (hg38) with default parameters. The alignment results were saved in BAM format. CUFFLINK-CUFFDIFF tools were used for expression analysis for RNA-seq data. The work flow of IPF high-throughput data processing to generate high confidence gene sets is given in Figure 3. For Micro-array gene data, GEO2R (http://www.ncbi.nlm.nih.gov/geo/geo2r/) tool containing R, Biobase46, GEOquery47 and limma48 packages was used with some basic changes in Rscript. Based on the information derived from the clustering of different gene sets with varying number of DEGs (detailed in the result section), set of feature genes was formed. The relative genome wide normalized expression values of these feature genes were used for vector transformational representation of each individual sample considered in this study. Features expression transformation into Z-score for each experimental dataset was done to bring the expression values on same scale irrespective of their platform and experimental design. The classification was implemented with 5-fold cross validation using LibSVM.
Identification of IPF specific TFs and miRNAs
DEGs having TF activity were identified. Sequences of DEGs were downloaded from Ensembl and were searched for homology in UniProt and AnimalTFDB 2.050 using BLASTX with the E-value threshold of 1E-05. Binding sites analysis was done using PROMO (TRANSFAC v8.3)51, CHIP-seq (ReMap)52 and manual curations. As the TFs activate the gene expression, positive expression correlation is expected between the TF-target gene pair. Z-score normalized expression values were used to find the correlation between TF and target genes. For selecting the significantly enriched categories, p-value cut off (0.05) with Benjamini-Hochberg test adjustment was applied.
miRNA expression analysis was done using four high throughput studies, mentioned above. Interestingly, so far, no small RNA-seq study has been done for miRNAs with respect to IPF. In this study the RNA-seq reads were utilized to detect the active miRNA regions of genome. The overlapped reads with miRNA precursor regions (taken from miRBase V21) were expected to express along with other coding genes. Such mapping reads were capable to provide the expression information of the precursor miRNAs, which in turn indicate the expression level of mature miRNAs. Differentially expressed miRNAs were selected using log2fold change (>=1) considering RPKM values in both conditions, whereas DE miRNAs from MA studies were selected using GEO2R (p-value<0.05). miRNAs which were having at-least two or more studies with same differential expression status were taken as final set of DEmiR. miRNAs from RNA-seq approach were preferred over MA in case of contradictory DE status (Supplementary Dataset 3). The filtered DEmiRs were analyzed for their target genes using TargetScan53 supplemented with experimentally validated interactions data from CLASH and CLIP-seq using miRTarBase version 6.054 while considering a negative expression correlation ("r"<=-0.7) for each miRNA-target. A work flow for miRNA analysis is provided in Supplementary Figure S8.
miRNA expression analysis was done using four high throughput studies, mentioned above. Interestingly, so far, no small RNA-seq study has been done for miRNAs with respect to IPF. In this study the RNA-seq reads were utilized to detect the active miRNA regions of genome. The overlapped reads with miRNA precursor regions (taken from miRBase V21) were expected to express along with other coding genes. Such mapping reads were capable to provide the expression information of the precursor miRNAs, which in turn indicate the expression level of mature miRNAs. Differentially expressed miRNAs were selected using log2fold change (>=1) considering RPKM values in both conditions, whereas DE miRNAs from MA studies were selected using GEO2R (p-value<0.05). miRNAs which were having at-least two or more studies with same differential expression status were taken as final set of DEmiR. miRNAs from RNA-seq approach were preferred over MA in case of contradictory DE status (Supplementary Dataset 3). The filtered DEmiRs were analyzed for their target genes using TargetScan53 supplemented with experimentally validated interactions data from CLASH and CLIP-seq using miRTarBase version 6.054 while considering a negative expression correlation ("r"<=-0.7) for each miRNA-target. A work flow for miRNA analysis is provided in Supplementary Figure S8.
IPF mediated regulatory network construction and motifs identification
RRegulatory relationships in form of miRNA-gene, miRNA-TF, TF-gene and TF-miRNA were considered for generation of regulatory network to unveil disease mechanism in IPF. Differentially expressed miRNAs, genes in IPF condition with respect to normal patient and TFs controlling both were retrieved for deciphering all such relationships. All types of interaction pairs were merged and unified to construct IPF-specific regulatory network. TF and miRNA regulatory network modules having various potential Feed-Forward Loops (FFLs) were derived at k-threshold of 3 using clique percolation algorithm, CFinder55. Potential FFLs are made of interconnected complete subgraph of TFs, differentially expressed miRNAs and genes. Variations in the type of potential FFL occur due to regulatory interaction between TFs and miRNAs. The work flow for the construction of TF-miRNA mediated regulatory network and statistics of three and four node FFLs for the three sets is illustrated in Supplementary Figure S9A and B, respectively.
IPF specific potential FFLs were evaluated for their significance in comparison to randomized networks. For this purpose, we compared real regulatory networks to randomized ones, preserving same degree as for the real one to estimate the probability that an FFL appears in the randomized networks. 1000 randomized networks were built, implementing edge swapping algorithm present in igraph56 R-package. The degree was kept same as for real regulatory network. All potential FFLs in each of the regulatory network were statistically analyzed for their significance while comparing to 1000 randomized regulatory networks. Only those FFLs were considered further for the depiction of regulatory mechanism in IPF which were found statistically significant at 5% level.
Network properties of built TF-miRNA derived regulatory networks were extracted as a consequence of their significance. Cytoscape version 3.157 was used for network visualization and analysis of various network parameters such as degree connectivity, betweenness and closeness centrality etc. Hub miRNAs and TFs were identified by sorting their outdegrees in descending order and top five hub components in each category were separated. Hub IPF genes were determined on the basis of indegree distribution.
IPF specific potential FFLs were evaluated for their significance in comparison to randomized networks. For this purpose, we compared real regulatory networks to randomized ones, preserving same degree as for the real one to estimate the probability that an FFL appears in the randomized networks. 1000 randomized networks were built, implementing edge swapping algorithm present in igraph56 R-package. The degree was kept same as for real regulatory network. All potential FFLs in each of the regulatory network were statistically analyzed for their significance while comparing to 1000 randomized regulatory networks. Only those FFLs were considered further for the depiction of regulatory mechanism in IPF which were found statistically significant at 5% level.
Network properties of built TF-miRNA derived regulatory networks were extracted as a consequence of their significance. Cytoscape version 3.157 was used for network visualization and analysis of various network parameters such as degree connectivity, betweenness and closeness centrality etc. Hub miRNAs and TFs were identified by sorting their outdegrees in descending order and top five hub components in each category were separated. Hub IPF genes were determined on the basis of indegree distribution.
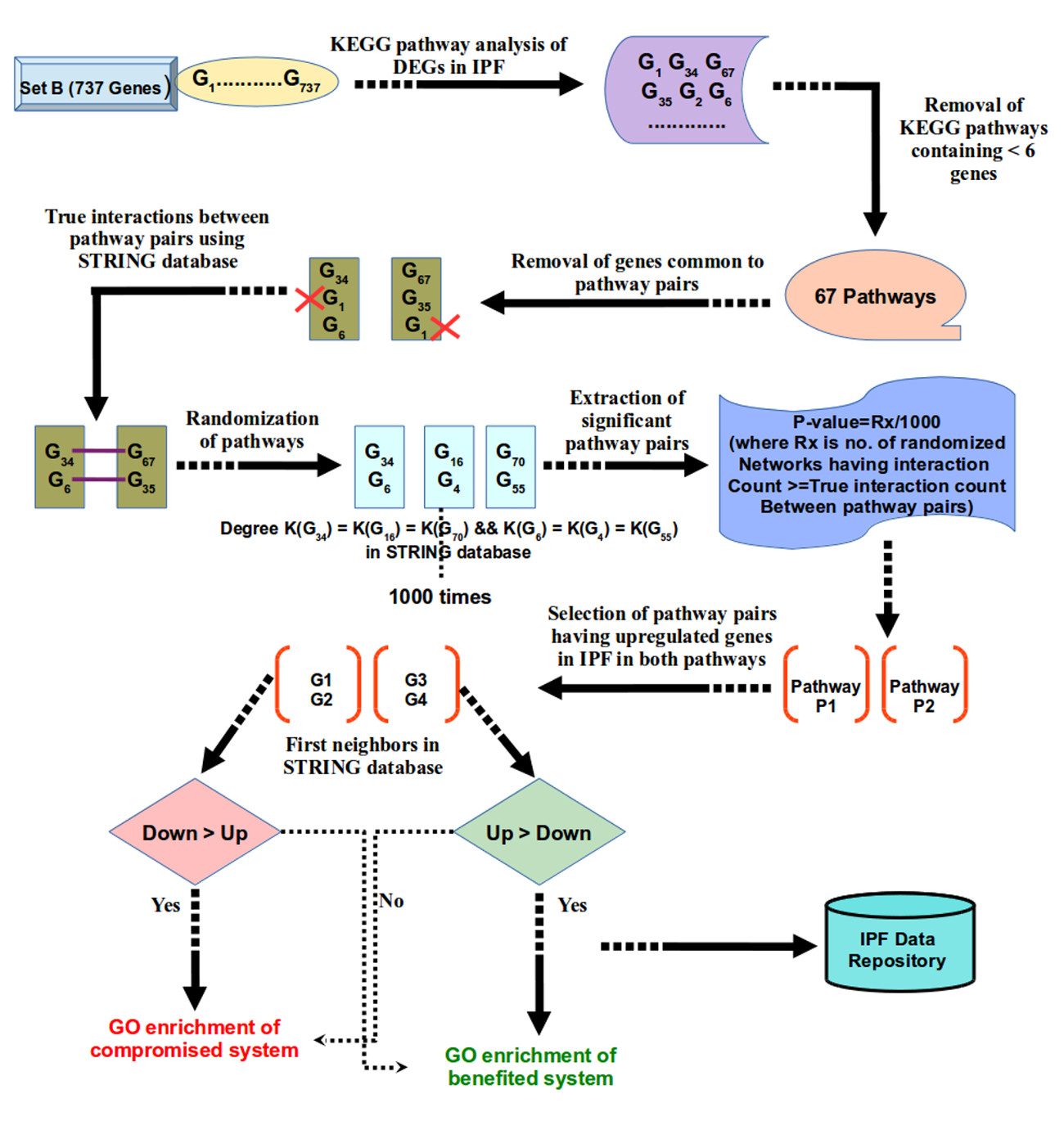
Pathway crosstalk analysis in IPF
Set B DEGs were utilized for pathway crosstalk network reconstruction. Pathways information related to such genes were retrieved from KEGG database. Human protein interaction data was downloaded from STRING v10 repository58 of functional protein interaction. A global pathway crosstalk approach was utilized to decipher IPF mediated pathway crosstalk59. To determine the background distribution, each pathway was randomized via shuffling of genes with genes having same degree in PPI database. Randomization process was performed 1000 times and protein interaction count was calculated for all randomized pathway pairs. Protein pairs which scored significant p-value against the random model pair occurrence were considered as cross talk pairs.
The obtained significant pathway pairs in crosstalk were filtered for those pairs having differentially up regulated genes in both pathways. First neighbors of these DEGs in STRING database containing confidence of 500 (covering most of the experimentally validated protein-protein interactions) were chosen for further analysis. Gene ontology enrichment analysis was done using GO-TermFinder60. Gene sets were extracted for benefited and compromised system chains due to pathway crosstalk in the disease condition. Detailed illustration representing pathway crosstalk network analysis pipeline in IPF is shown in Figure 4. The full method details have been made available in the supplementary methods.
The obtained significant pathway pairs in crosstalk were filtered for those pairs having differentially up regulated genes in both pathways. First neighbors of these DEGs in STRING database containing confidence of 500 (covering most of the experimentally validated protein-protein interactions) were chosen for further analysis. Gene ontology enrichment analysis was done using GO-TermFinder60. Gene sets were extracted for benefited and compromised system chains due to pathway crosstalk in the disease condition. Detailed illustration representing pathway crosstalk network analysis pipeline in IPF is shown in Figure 4. The full method details have been made available in the supplementary methods.
IPF Information Repository
The portal is implemented using advance libraries of HTML5. Portal homepage and background pages are developed in HTML5/CSS, JQuery/JavaScript with support of Bootstrap packages. Portal work flow is described in Supplementary Figure S10. Background connection is established by PHP version 5.2 with PERL version 5.18 and shell scripting. MySQL version 5.7 database connection is established through the PHP. The database provides the facility for IPF sample identification from expression data, crosstalk and network analysis and can be searched for genes, miRNAs and important pathways. All the interaction can be visualized in selective mode. Common target analysis is also activated for miRNAs which can be extended to the network. All the network elements can be selected for pathways and functional enrichment analysis. The analysis can be done in comparative mode. The network relationships between nodes and edges are structured into JSON file via in-house built PERL script. A set of full network JSON file is loaded into D3, a web based visualization open library. Network properties are represented into html tables using JavaScript for gene regulatory and protein-protein interaction networks. Query sent by a user from HTML pages is processed through PHP in the background and retrieves results of respective sections (like search, network, classification of IPF samples etc.).
The portal is implemented using advance libraries of HTML5. Portal homepage and background pages are developed in HTML5/CSS, JQuery/JavaScript with support of Bootstrap packages. Portal work flow is described in Supplementary Figure S10. Background connection is established by PHP version 5.2 with PERL version 5.18 and shell scripting. MySQL version 5.7 database connection is established through the PHP. The database provides the facility for IPF sample identification from expression data, crosstalk and network analysis and can be searched for genes, miRNAs and important pathways. All the interaction can be visualized in selective mode. Common target analysis is also activated for miRNAs which can be extended to the network. All the network elements can be selected for pathways and functional enrichment analysis. The analysis can be done in comparative mode. The network relationships between nodes and edges are structured into JSON file via in-house built PERL script. A set of full network JSON file is loaded into D3, a web based visualization open library. Network properties are represented into html tables using JavaScript for gene regulatory and protein-protein interaction networks. Query sent by a user from HTML pages is processed through PHP in the background and retrieves results of respective sections (like search, network, classification of IPF samples etc.).
IPF Information portal implementation
Among several network based approaches, a widely used algorithm of Random walk with restart [51], was utilized to rank differentially expressed genes of IPF. The algorithm considers whole network traversal through random walk. In this study, due to very limited availability of disease associated genes so far reported to diagnose and identify IPF, we have selected the eight disease genes obtained from a database of human diseases and their associated genes entitled as DisGeNET [52] as starting seed points. Random walk with restart (RWR) considers the adjacency matrix of the protein interaction network and transition matrix is calculated characterizing the probability of transition from one node to other. Every step of random walk assigned a probability score to each network node deciphering its chance to travel the node. Until the convergence of probabilities, algorithm updates the score utilizing the following equation:
where, Pt+1 is probability of random walk at time "t", W' is transpose of adjacency matrix of PPI network taken in account, P0 is initial probability and "r" denotes the restart probability threshold of random walk at each step of traversal. Nodes having highest probability scores are considered as in close proximity of all disease genes of IPF and depicted as candidate IPF genes. This all helps in selecting the target genes for drug discovery.
Among several network based approaches, a widely used algorithm of Random walk with restart [51], was utilized to rank differentially expressed genes of IPF. The algorithm considers whole network traversal through random walk. In this study, due to very limited availability of disease associated genes so far reported to diagnose and identify IPF, we have selected the eight disease genes obtained from a database of human diseases and their associated genes entitled as DisGeNET [52] as starting seed points. Random walk with restart (RWR) considers the adjacency matrix of the protein interaction network and transition matrix is calculated characterizing the probability of transition from one node to other. Every step of random walk assigned a probability score to each network node deciphering its chance to travel the node. Until the convergence of probabilities, algorithm updates the score utilizing the following equation:
where, Pt+1 is probability of random walk at time "t", W' is transpose of adjacency matrix of PPI network taken in account, P0 is initial probability and "r" denotes the restart probability threshold of random walk at each step of traversal. Nodes having highest probability scores are considered as in close proximity of all disease genes of IPF and depicted as candidate IPF genes. This all helps in selecting the target genes for drug discovery.

After analysis of a large volume of high throughput genomic data, a set of 39 differentially expressed genes (DEGs) emerged critical to distinguish IPF. Experimentally validated and high confidence data with multiple evidences were used to reconstruct the system model for IPF, incorporating various PPI and regulatory interactions. Careful analysis of the networks identified several critical potential FFLs as well as compromised and benefited routes in various pathways, marking the phenotype of IPF. A system of serially connected five genes, eight TFs and nine miRNAs appeared exclusive to IPF, which could be promising for therapeutic interventions.
In future, the expression of associated miRNAs in this system could be controlled in order to verify their potential role as therapeutic targets for IPF to design therapeutic agents. Finally, this study has generated a state-of-the-art database on IPF where the involved components can be analyzed and visualized in a highly informative manner. This database would be very useful for any molecular systems research on IPF.