

Explainable Generative Deep Learning for plant transcription factor binding sites discovery
Source code/Standalone version of the program is hosted at the GitLab
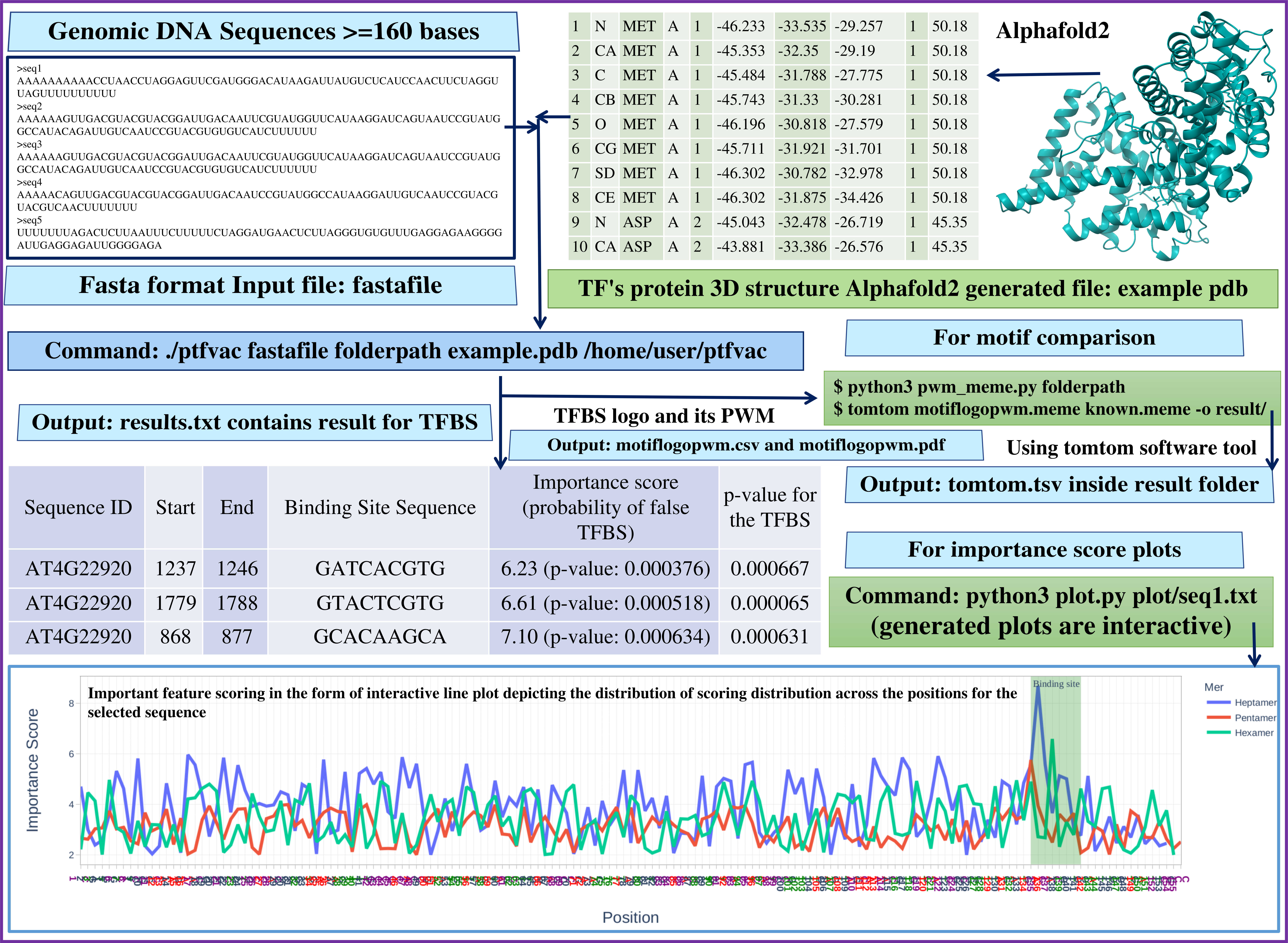
Figure: Implementation of the PTF-Vāc standalone system.
Running the standalone program
Create & activate virtual environment and install other required dependencies
conda create -n ptfvac python=3.8.10 -y
conda activate ptfvac
unzip software.zip
chmod a+x INSTALL
./INSTALL (This step will take some time to install all dependencies in one go)
cd deeptfactor
sh meme.sh (This step will take some time to install MEME suite)
Note: Always use Alphafold2 generated TF pdb file only (An example is provided). This standalone version was developed in Linux, and has been tested in Ubuntu 20.04 with Python 3.8.10.
Input
fastafile = File containing fasta sequences.
example.pdb = Alphafold2 generated example pdb file.
Running script
To detect the TF binding sites, In parent directory execute following command:
Usage: ./ptfvac "fastafile" "folderpath" "Alphafold generated PDB file" "folderpath of deeptfactor inside PTF-Vac"
eg: ./ptfvac fastafile folderpath example.pdb /home/user/ptfvac
See the "Output" section below for the files generated by this command.
To generate line plot, execute the following command:
python3 plot.py plot/seq1.txt (filename) (generated plots are interactive)
For comparison between motif identified by PTF-Vac and already known motif (In MEME format, an example is provided "known.meme")
Step 1: Convert PTF-Vac motif to MEME format
python3 pwm_meme.py folderpath
Run TOMTOM
Step 2: tomtom motiflogopwm.meme known.meme -o result/folder
Output
results.txt = The TF binding site information regarding the binding site sequence and it's start and end coordinates in the sequence. The results also provides the binding site sequence importance score, its binomial test based p-value, and hypergeometric test based p-value.
motiflogopwm.csv = Position weight matrix of binding site sequences.
motiflogopwm.pdf = Sequence logo of binding site sequences.
plots = A folder containing importance score plot.
Note
For HADDOCK implementation see README_HADDOCK.md file for more details.
>seq_1
UGAUAAACAAAGUGUGUAACAUCACCUCAUCUACAUGUGUGAUUUUUUUUUUGAAUAUAGACAACUUUUUAGUCAGAGUUUACAUGAGUUUUCACCUAAUUUGUGGUUUAAUUACACCGCAUAUUU
GCCCAAUUUAGUGAGUAUAGUGAGUUUCUGUAGAGAAGCUCAUCUUAGAAUUAUUCAUGUAUUCCACUACUAAA
>seq_2
AGAUCUACAAGAGAAGAUAAGUUUGAGGCAAAUUCGAGAUCUGGAAGCUGGUUUUCUCUUUACAAAUAACACUAACCCUACCAUCAAAUCAAGAAAGGAGGCUUUGAACAAAUAGCUUGAUUGAAG
UAUGAAGUGGCUCGGUGGGCGACGAUGACGGGCGAGCUCCGGCGAGGGCCUGGGGGCCUGAGCGACGAUGACGG
>seq_3
AACGGGUCGUGCCGGCACGGCCCACGAGCGGGCGUGCCGUGCCGUUCCUGGGCCGGCUACAGUGCUGCCGUGCUCGGGCCGGCACGCCUUGGCCCGGCCCAUUUGGCCAGGUAUACUAGUCGGCUC
CAGUCCUCCUCCCCCCAGCGACCUAAGCCGCCACCGCCCUCGCCGCGCUACCGCCAGCGCCGCCUGCCGUCCCU
>seq_4
UUUUCCCCUUGAUUUUAGGGUUAGGGUUUCAUGAUUUGGGGAAAAAUUUGGGAUCUUACUGUAGCUAGGGUUUCGGUUCUUGGGGAUUUGUCUGAGAUUUGCAUGAACUUUUGCUUUCCCCUUCUU
CUAAUUUCUUCAACCCGAAACCCUAGAAGGCCUAAUUCCAUUUCUUAUAUUUCGGGAUUGCAUGAUUUGGGCUU
>seq_5
CCGGACGAUAGCAAGCGCUGGCAGUAGAGUAGGCUAGAGUCAUGAGUCUGAGUCAUGCUGGCUUUAUAUAGACAAAAAAUGGUACUACACACAAAUGAAAUUUCUAGCAAAAAUAAUCAAUGCACU
UUCCUUGAUUACACACCAACUUUAUGUAUAUAUAGGCUGGAAUAAUCCAUUGUGCAUGUACAUGAAUAUAGAUU
>seq_Ath
UGAUAAACAAAGUGUGUAACAUCACCUCAUCUACAUGUGUGAUUUUUUUUUUGAAUAUAGACAACUUUUUAGUCAGAGUUUACAUGAGUUUUCACCUAAUUUGUGGUUUAAUUACACCGCAUAUUU
GCCCAAUUUAGUGAGUAUAGUGAGUUUCUGUAGAGAAGCUCAUCUUAGAAUUAUUCAUGUAUUCCACUACUAAAAGAUCUACAAGAGAAGAUAAGUUUGAGGCAAAUUCGAGAUCUGGAAGCUGGU
UUUCUCUUUACAAAUAACACUAACCCUACCAUCAAAUCAAGAAAGGAGGCUUUGAACAAAUAGCUUGAUUGAAGUAUGAAGUGGCUCGGUGGGCGACGAUGACGGGCGAGCUCCGGCGAGGGCCUG
GGGGCCUGAGCGACGAUGACGGAACGGGUCGUGCCGGCACGGCCCACGAGCGGGCGUGCCGUGCCGUUCCUGGGCCGGCUACAGUGCUGCCGUGCUCGGGCCGGCACGCCUUGGCCCGGCCCAUUU
GGCCAGGUAUACUAGUCGGCUCCAGUCCUCCUCCCCCCAGCGACCUAAGCCGCCACCGCCCUCGCCGCGCUACCGCCAGCGCCGCCUGCCGUCCCUUUUUCCCCUUGAUUUUAGGGUUAGGGUUUC
AUGAUUUGGGGAAAAAUUUGGGAUCUUACUGUAGCUAGGGUUUCGGUUCUUGGGGAUUUGUCUGAGAUUUGCAUGAACUUUUGCUUUCCCCUUCUUCUAAUUUCUUCAACCCGAAACCCUAGAAGG
CCUAAUUCCAUUUCUUAUAUUUCGGGAUUGCAUGAUUUGGGCUUCCGGACGAUAGCAAGCGCUGGCAGUAGAGUAGGCUAGAGUCAUGAGUCUGAGUCAUGCUGGCUUUAUAUAGACAAAAAAUGG
UACUACACACAAAUGAAAUUUCUAGCAAAAAUAAUCAAUGCACUUUCCUUGAUUACACACCAACUUUAUGUAUAUAUAGGCUGGAAUAAUCCAUUGUGCAUGUACAUGAAUAUAGAUU
B: Bimodal CNN
@SEQ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTTT
+
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC65
chr1 0 100 abcd 255 +
chr1 10 125 abcd 255 -

