

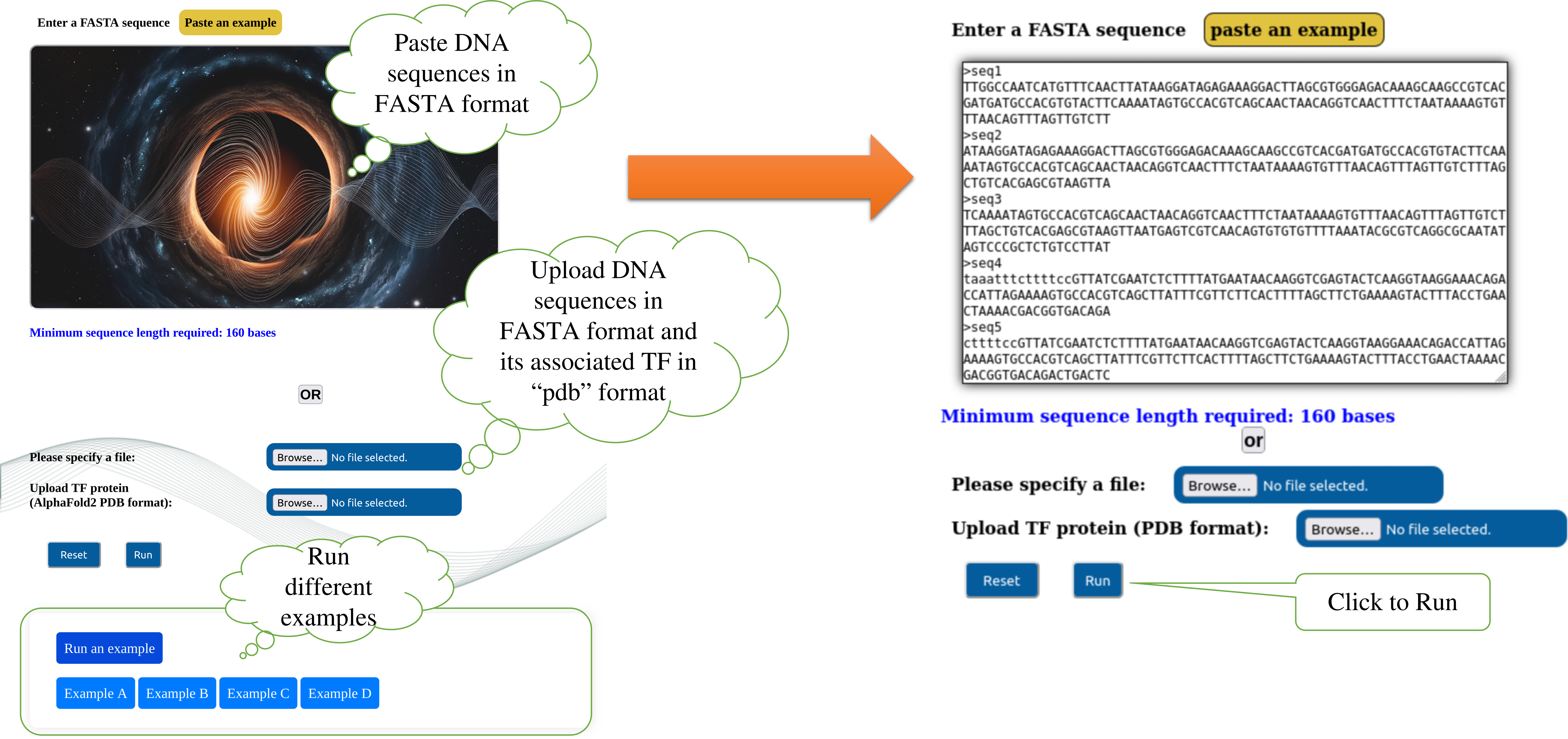
Step1: a) Input data box where the user can either paste or upload the input files. Inputs are genomic DNA sequence in FASTA format with unique sequence identifier and 3D TF structure generated from Alphafold2 in pdb format. In the first part user can only paste or load the example while the pdb file will be copied to the user destination page by default. Here, ABF2 TF is being used as by default pdb file. In the upload section, user needs to upload both genomic DNA sequence and pdb files. It either of the files were not uploaded and submitted, the page will throw error regarding the same. b) In the right side part, user can select their species, TF, and target gene whose promoter to be analysed from the dropdown options provided (For results output specific to this part see Step 11).

Step2: Upon submission, a loading page will appear until the results are ready.

Step 3: The binding site information is represented in the form of table. Table consists of information regarding the binding site sequence and it's start and end coordinates in the sequence. The results also provides the binding site sequence importance score, its binomial test based p-value, and hypergeometric test based p-value. User can also download the result in the tabular format. An option is alo provided to perform molecular docking of TFBS and the corresponding protein using HADDOCK3 (see Step 9 and 10). User can also visualize 3D structure of protein with its sequence and sequence logo of the binding sites, detailed in the next step.
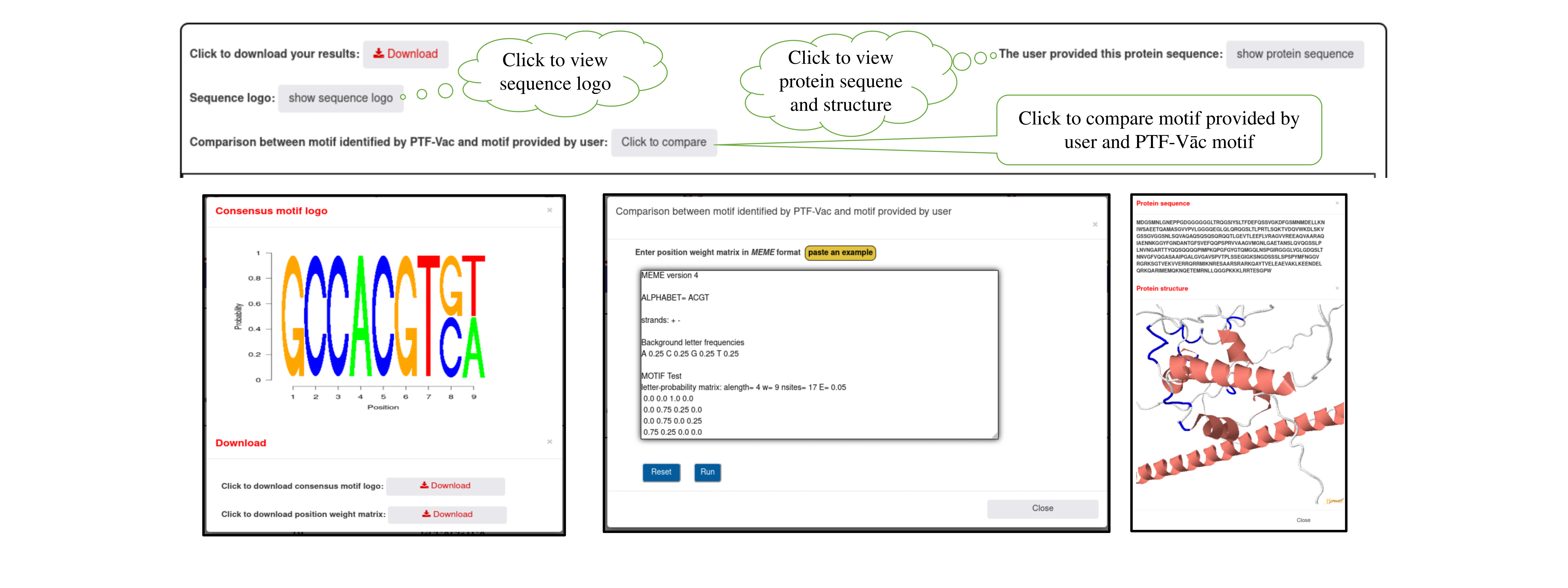
Step 4: User can visualize 3D structure of protein with its amino acid sequence. Visualization of 3D structure is powered by JSmol. By clicking "show sequence logo", sequence logo of the binding sites will be displayed, which can downloaded in png and PWM format. This PWM can be benchmarked with the user's PWM, upon clicking "click to compare", detailed in the next step.
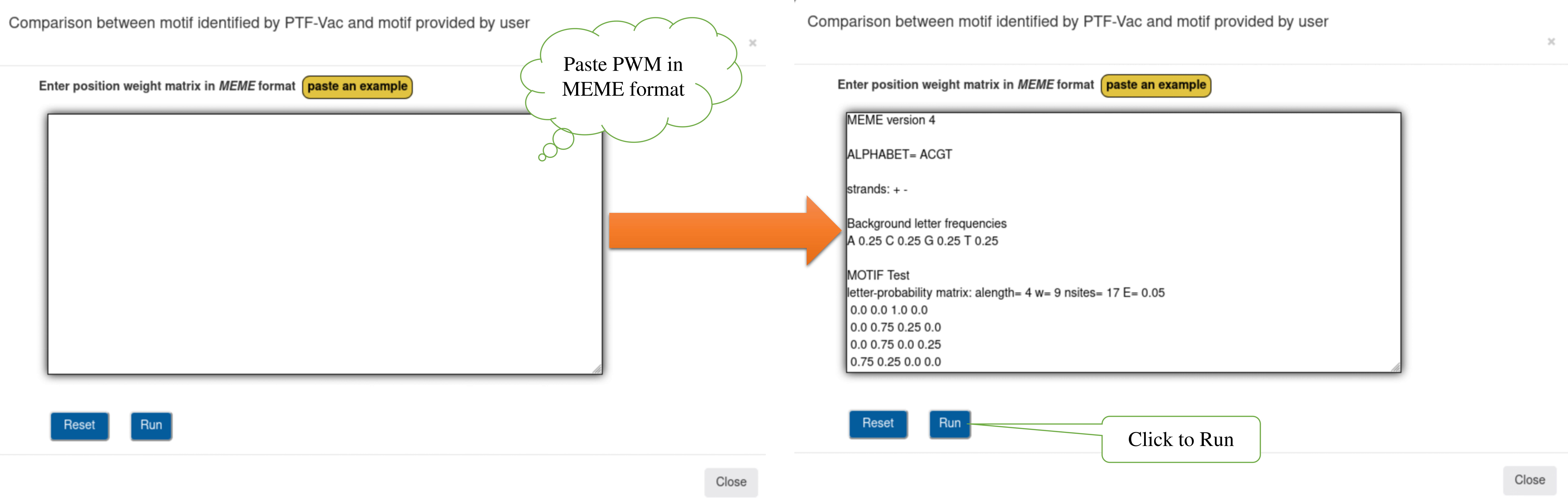
Step 5: With integration of TOMTOM in the backend, users can able to compare the PTF-Vac generated motif's PWM with their own choice of PWM, enabling a comparative analysis of binding motifs across different datasets or experimental conditions. Input is PWM in MEME format, user can load or paste the example.

Step 6: Output result of TOMTOM for better interpretation of result. User can also download the result by clicking the download button.
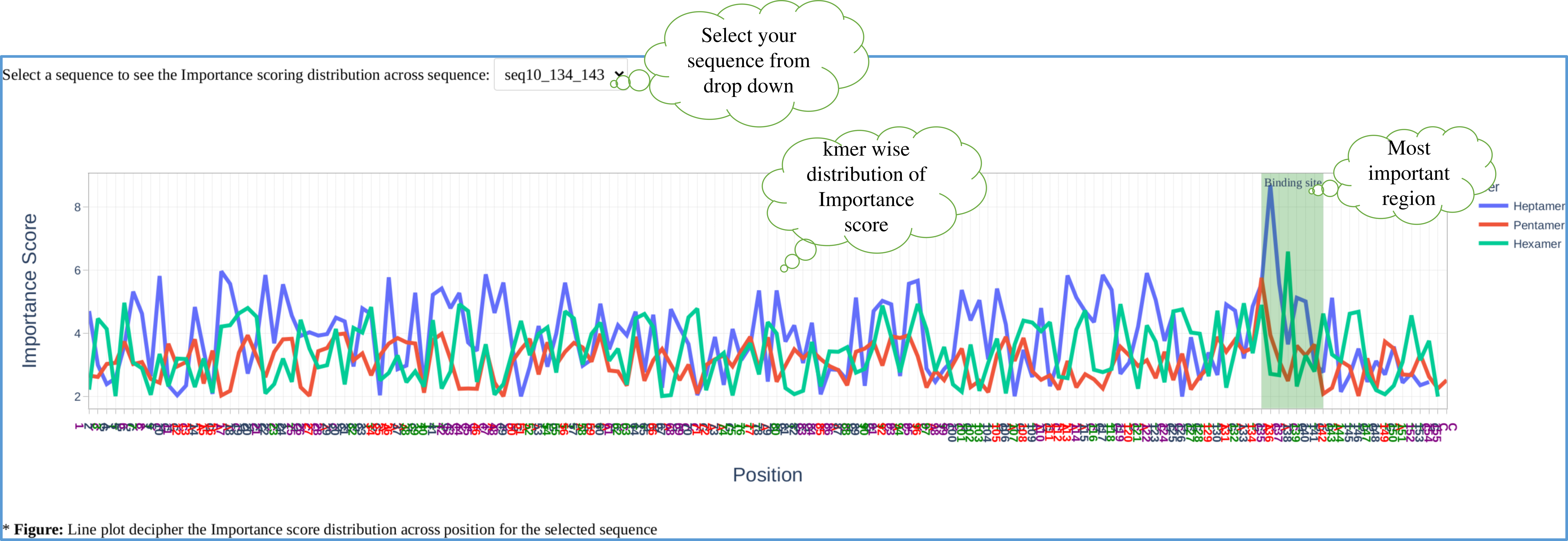
Step 7: Important feature scoring is implemented and is represented in the form of interactive line plot depicting the distribution of scoring distribution across the positions for the selected sequence. PTF-Vac uses Grad-CAM, to highlight important regions in input sequences. The importance score distribution shows which nucleotide positions most influence TFBS identifications. This helps to interpret the model's decisions and identify biologically relevant binding patterns. Here the short kmers for example, Heptamers, Hexamers, and Pentamers are all analyzed to show which specific patterns or sub-sequences play a key role in TF binding. This visualization helps users to understand: where the core binding motif lies, which parts of the sequence are most critical for TF recognition, and whether the model\u2019s TF motif identification matches known binding sites.
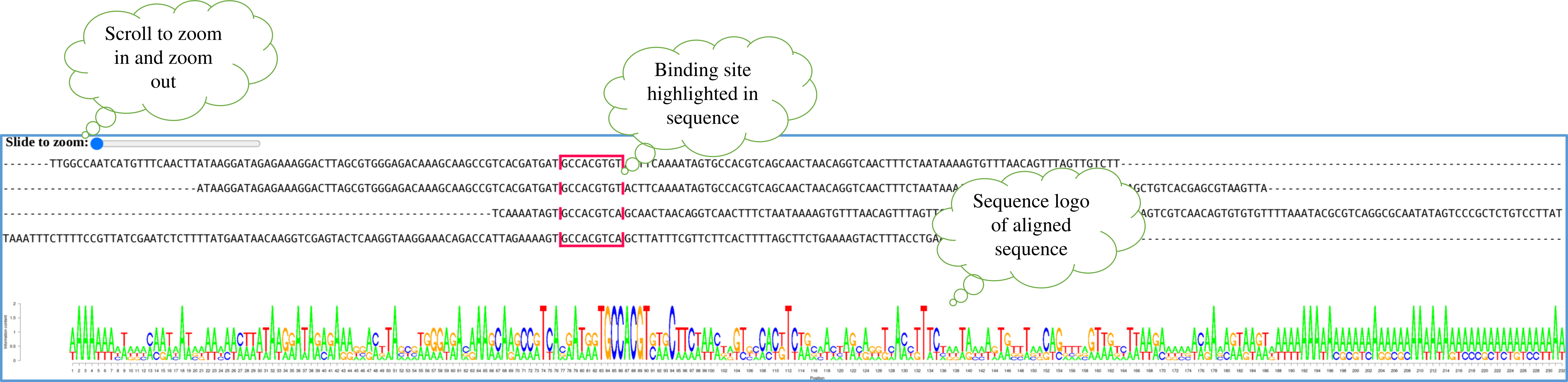
Step 8: The binding site is highlighted among the sequences and a motif logo is also built from consensus.
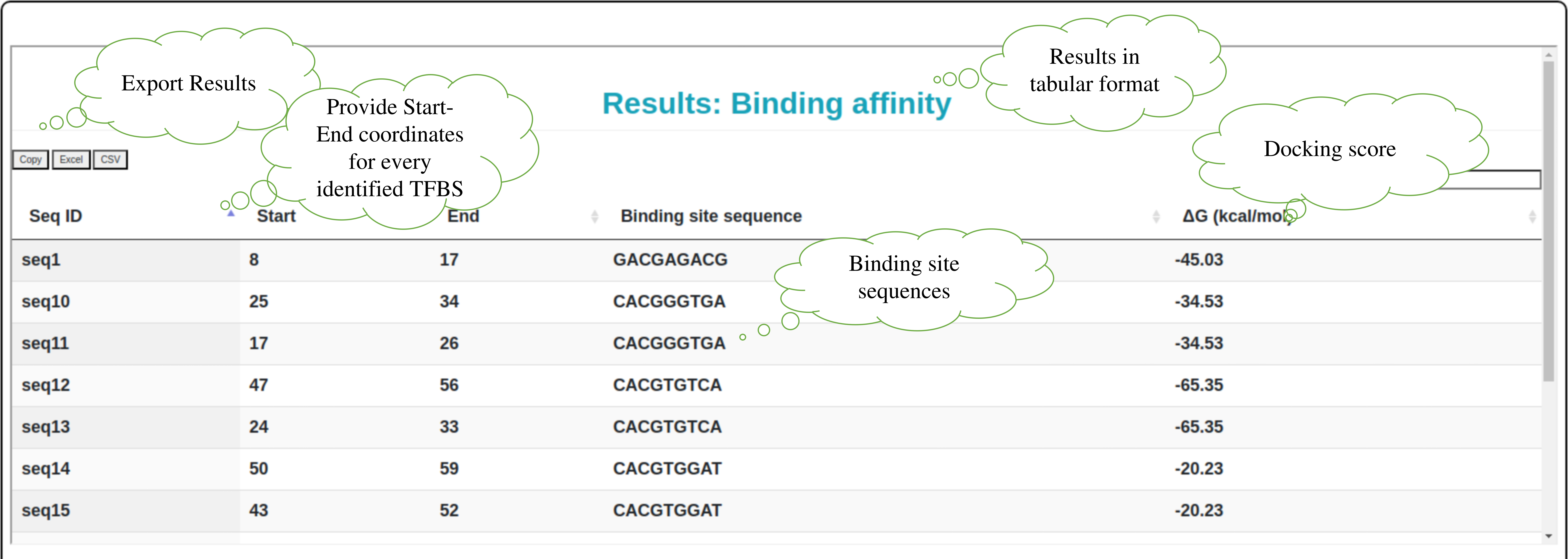
Step 9: Snapshot providing description of molecular docking analysis using HADDOCK3 output in tabular form which includes binding affinity between TF-3D structure and its binding sites.
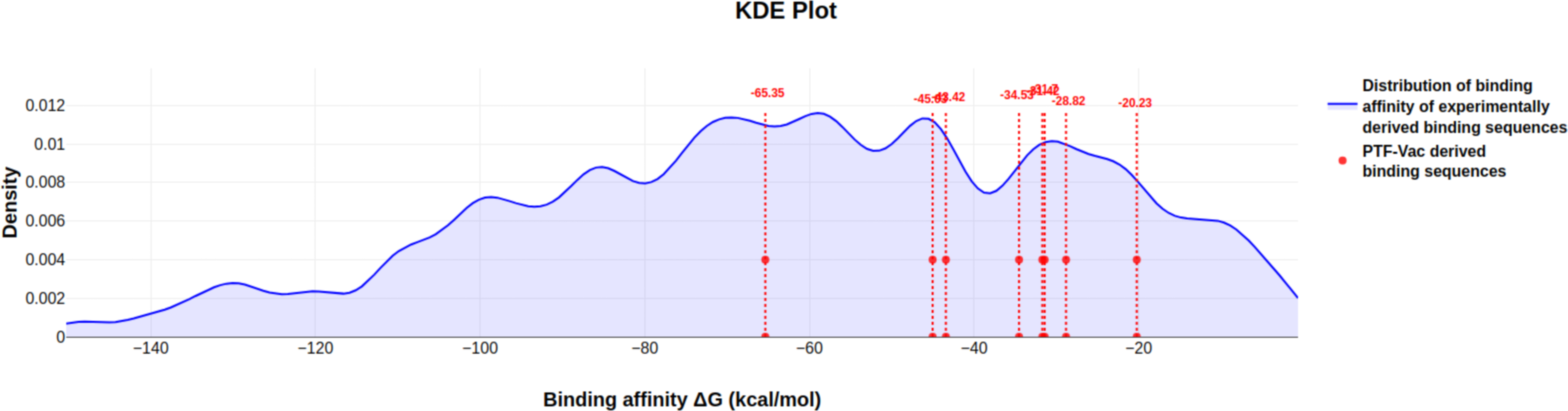
Step 10: Snapshot depicting KDEplot of binding affinity values from molecular docking analysis for the set of 563 TF-3D structure and their binding sites.
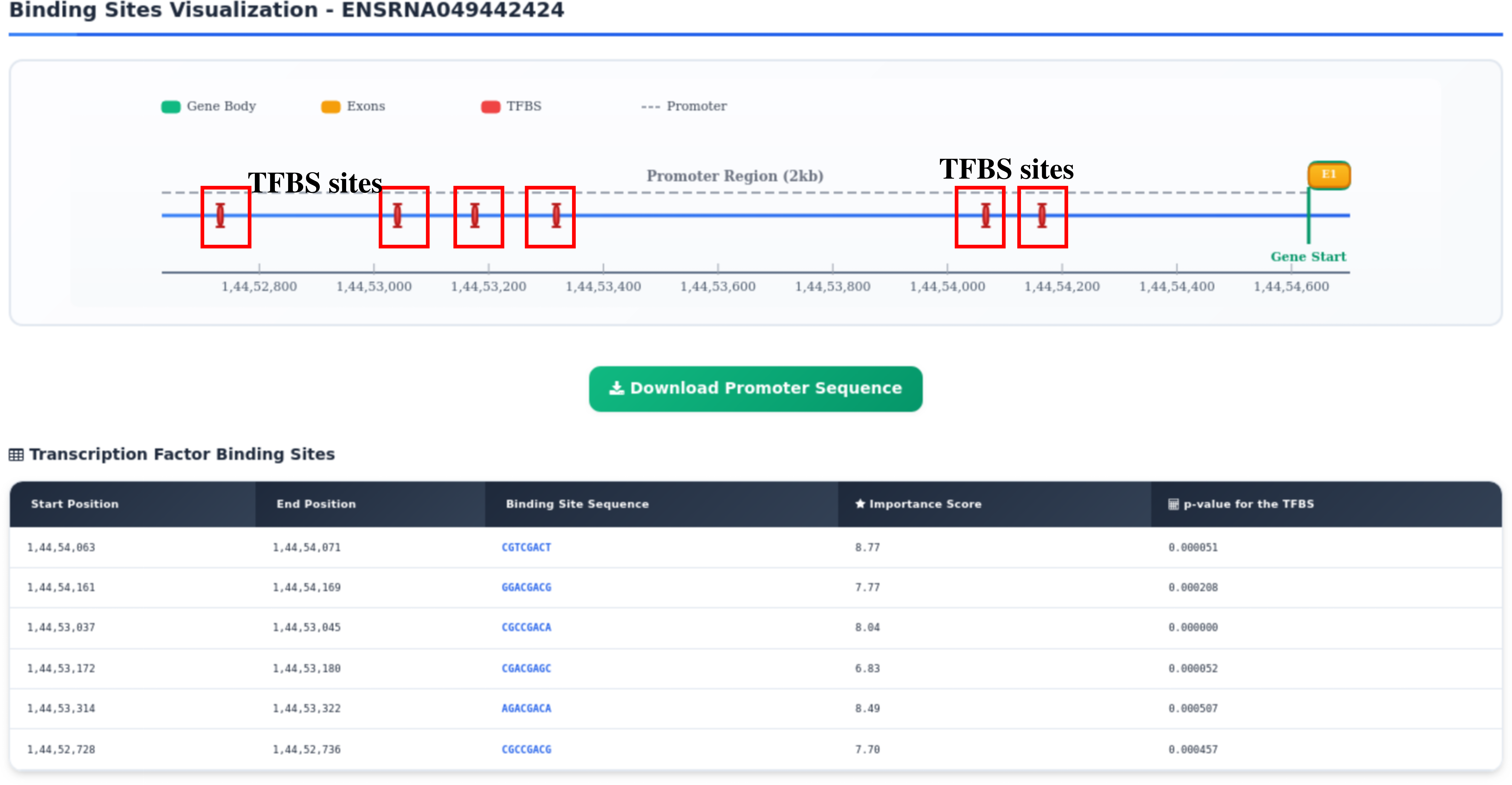
Step 11: Results are presented in the form of gene model annotated from GFF3 file with TFBS sites highlighted on it.
