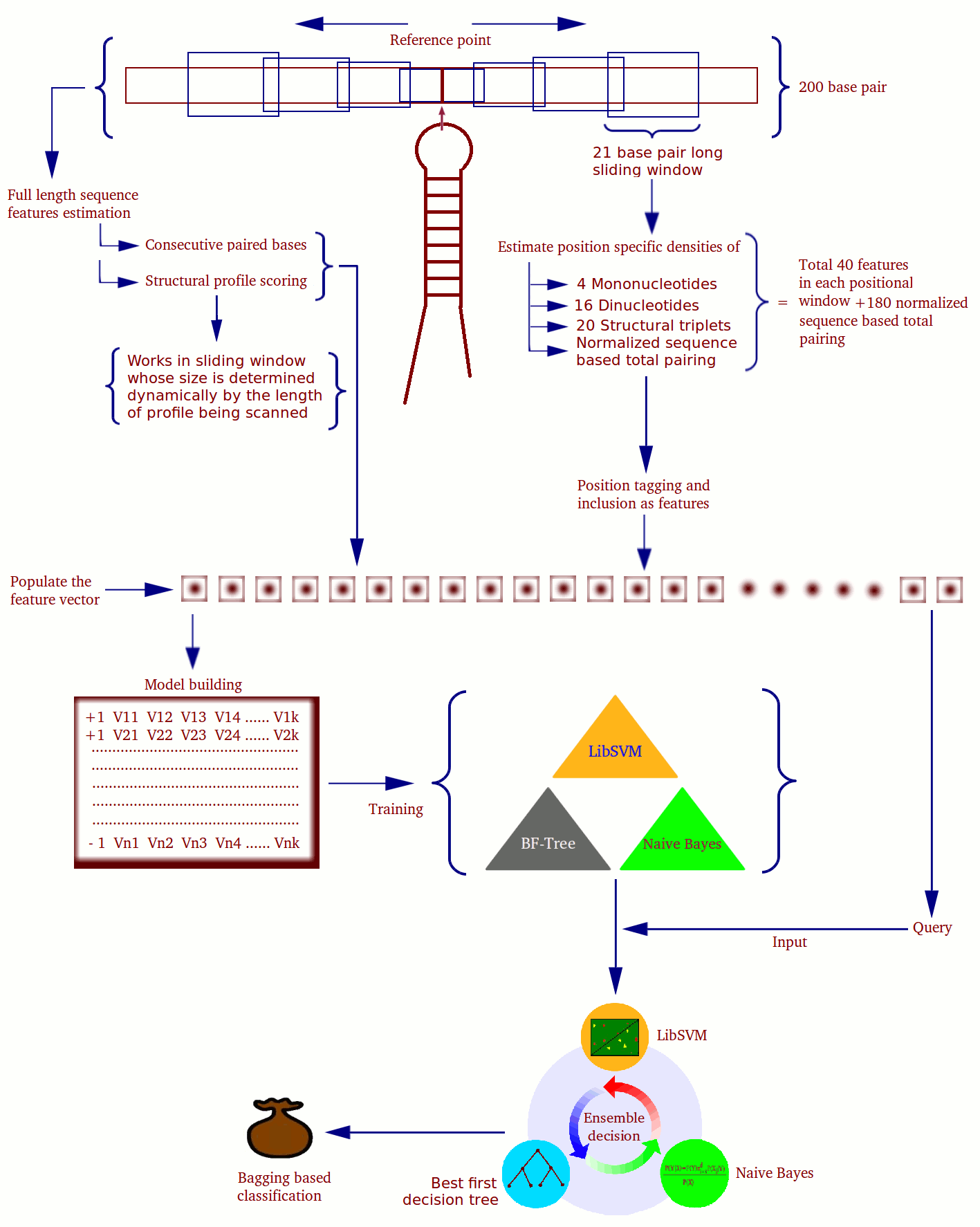
|
Web-server:
The web-server for miR-BAG is developed in HTML and PHP web technologies on Linux (Fedora). All the back-end programs are developed in java and perl along with Linux shell scripting. Web server gives the option of concurrency to user but it is recommended that user should download the stand-alone for miR-BAG.
miR-BAG server provide two choices to the user, one module is for simple sequence scan and another is for miRNA identification from Next Generation Sequencing (NGS) data. Following parameters should be kept in mind before submitting sequences to miR-BAG
To run miR-BAG Sequence-scan-module
-
The input file format should be in fasta format.
User can upload a file or paste the sequence in Paste sequence text area. An example is given in the web server for correct input.

-
The minimum length of the sequence(s) must be 200 bases while the maximum length should be upto 300 bases.
-
User can submit multiple sequences but it is advisable to submit only one sequence at a time in web server for fast output.
-
Select the model and number of processors from the corresponding drop down lists.
-
The output consists of sequence ID, start and end position of the best precursor sequence and its classification score. miR-BAG uses bagging with three classifiers, and the scoring scheme depicts that sequence(s) having score 0.3 is considered as positive miRNA candidate by one classifier, sequence(s) having score 0.6 is considered as positive miRNA candidate by two classifiers and sequence(s) having score 1.0 is considered as positive miRNA candidate by all three classifiers.
Use of filtering options
It seems that while working with sequence length of exactly 200 bases the output is given instantly by miR-BAG. But if user submits a sequence of length above 200 bases then miR-BAG has to work on sliding fragments of 200 bases. If user has submitted a sequence of length 300 bases then miR-BAG has to compute the data on 100 fragments repeatedly. In that case one sequence of lenght 300 bases is eqivalent to 100 sequences of length 200 bases for miR-BAG. This creation of fragments and computation of various features on these fragments takes a lot of time. User can reduce the number of fragments by selecting different filtering options. Six filtering options are given in the web-server of miR-BAG. User can click the particular checkbox of desired filter, then user has to enter the minimum cut-off value. The range for each filter is from the minimum cut-off value entered by the user to the highest value of that filter. Different statistics for each filtering option are given in the hyperlink in front of each filtering option.
To run miR-BAG NGS-module
-
The data should be provided in a tab separated format which includes the reads and their count separated by a tab.
To convert fastq file to a tab separated read file download the program available on webpage.

-
The data submitted by user will be mapped on pre-miRNA, non coding RNA sequences and mRNA sequences and all those reads which will not map on any of these sequences will be considered for analysis.
-
The remaining reads will be mapped on genome and sequence of 250 bases will be extracted from the genomic sequences of chosen species, which will be used for classification by miR-BAG.
Note: Currently on webserver only human model is given, to analyze other models please download the stand-alone. The mapping and final prediction might take some time in web server therefore it is highly recommended to install stand-alone on local system with Linux O.S.
|
|
|
|
Copyright © 2012, CSIR-Institute of Himalayan Bioresource Technology.
Developed & Maintained by Heikham Russiachand Singh, SCBB, Biotech Division
|